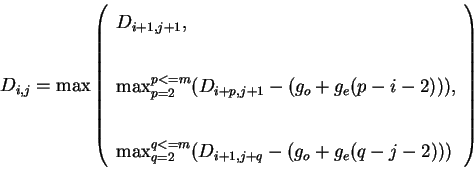| advertisement: compare things at compare-stuff.com! |
The first application of dynamic programming to biological sequence alignment (both DNA and protein) was by Needleman and Wunschneedleman:wunsch. This and related algorithms have been in use since then for the detection of similarities (see Section 1.4.1) and the alignment of sequence information from protein families.
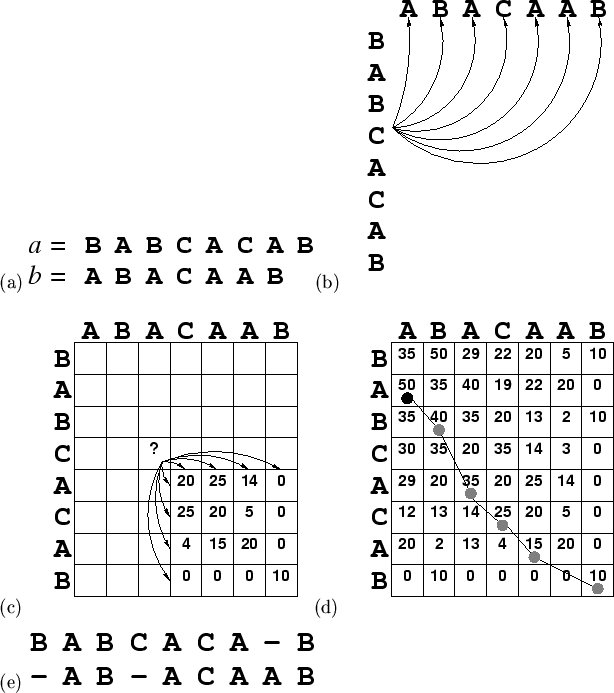
Figure 1.1(a) shows two short (imaginary) sequences we wish to
align: sequence ![]() = BABCACAB, sequence
= BABCACAB, sequence ![]() = BCCAAB. Residues
= BCCAAB. Residues ![]() and
and
![]() in sequences
in sequences ![]() and
and ![]() are denoted by
are denoted by ![]() and
and ![]() respectively.
Similarly their lengths are denoted
respectively.
Similarly their lengths are denoted ![]() and
and ![]() . The objective of any
alignment algorithm is to maximise the alignment of similar or identical
residues in
. The objective of any
alignment algorithm is to maximise the alignment of similar or identical
residues in ![]() with
with ![]() , equivalent to
, equivalent to ![]() along the path of
the alignment.
along the path of
the alignment. ![]() is best explained as how `happy' residue
is best explained as how `happy' residue ![]() is
aligned against
is
aligned against ![]() .
.
 |
In sequence comparison, the value of ![]() is looked up in a matrix
is looked up in a matrix ![]() containing the log-likelihoods of all substitutions between each of the 20
amino acids (
containing the log-likelihoods of all substitutions between each of the 20
amino acids (
![]() ).
). ![]() is approximated from the
frequencies of substitutions observed in sequence
alignments[Barker & Dayhoff, 1972,Dayhoff et al.,
1978,Henikoff & Henikoff, 1992]. Thus
is approximated from the
frequencies of substitutions observed in sequence
alignments[Barker & Dayhoff, 1972,Dayhoff et al.,
1978,Henikoff & Henikoff, 1992]. Thus
![]() and when
and when ![]() ,
, ![]() gives a high
positive value. Smaller or negative values of
gives a high
positive value. Smaller or negative values of ![]() occur when the
amino acids
occur when the
amino acids ![]() and
and ![]() are less alike in terms of observed
substitutions.
are less alike in terms of observed
substitutions.
Gapless alignment of sequence ![]() and
and ![]() would simply involve sliding one
sequence along the other and outputting the alignment where
would simply involve sliding one
sequence along the other and outputting the alignment where ![]() is maximal, involving a number calculations only linearly dependent upon
the sequence lengths. The allowance of gaps requires that each
is maximal, involving a number calculations only linearly dependent upon
the sequence lengths. The allowance of gaps requires that each ![]() must
be `tested' against
must
be `tested' against ![]() . Figure 1.1(b) illustrates the number
of possibilities which exist in the alignment of residue
. Figure 1.1(b) illustrates the number
of possibilities which exist in the alignment of residue ![]() (C) with
sequence
(C) with
sequence ![]() . There are
. There are ![]() such comparisons in total. Thus
gapped alignments involve calculations scaling approximately to the square
of number of residues.
such comparisons in total. Thus
gapped alignments involve calculations scaling approximately to the square
of number of residues.
The dynamic programming algorithm finds the optimal alignment through the
construction of the ![]() matrix
matrix ![]() . The values of
. The values of
![]() are filled in dynamically starting from the bottom right-hand
corner as follows:
are filled in dynamically starting from the bottom right-hand
corner as follows:
When complete, the highest value from the top and left edges of the matrix
![]() is found (marked with a black filled circle in Figure 1.1(d))
and the path which resulted in this score is traced back in reverse to
generate the alignment, shown in Figure 1.1(e). A relatively
minor modification to this algorithm was developed by Smith and
Waterman[Smith & Waterman, 1981] to locate the best local alignments between
two sequences. Local alignments are not required to include the termini of
either sequence, as in the Needleman and Wunschneedleman:wunsch
method which is a global alignment.
is found (marked with a black filled circle in Figure 1.1(d))
and the path which resulted in this score is traced back in reverse to
generate the alignment, shown in Figure 1.1(e). A relatively
minor modification to this algorithm was developed by Smith and
Waterman[Smith & Waterman, 1981] to locate the best local alignments between
two sequences. Local alignments are not required to include the termini of
either sequence, as in the Needleman and Wunschneedleman:wunsch
method which is a global alignment.