| advertisement: compare things at compare-stuff.com! |
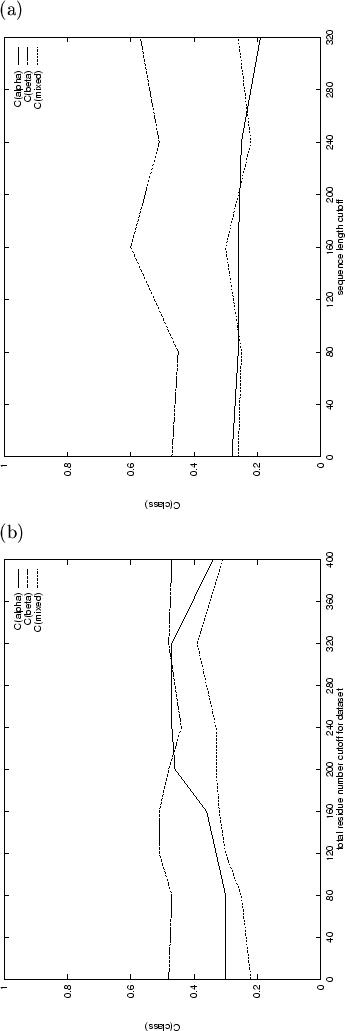
The composition vectors of short sequences are susceptible to large changes
as the result of just a few amino acid substitutions. The structural class
information content of these vectors is therefore unstable and is likely to
render predictions less reliable. To explore this, Results 8 to 11 in
Table 3.1 have been obtained using datasets with minimum
sequence length cutoffs of between 80 and 320 residues. Since the
structural class composition of the datasets is not constant, the most
suitable comparison between predictions is made using the Matthews
correlation coefficients for each class, which are plotted in
Figure 3.1(a). Improvements in ![]() are consistent,
however the predictions for the mainly-
are consistent,
however the predictions for the mainly-![]() and mixed-
and mixed-![]() protein domains are slightly worse using longer sequences. The use of
sequence length-filtered datasets greatly improves the accuracy of
predicting the presence or absence of
protein domains are slightly worse using longer sequences. The use of
sequence length-filtered datasets greatly improves the accuracy of
predicting the presence or absence of ![]() -helix (hence the high value
for
-helix (hence the high value
for ![]() ) but is not so good at distinguishing between the two
) but is not so good at distinguishing between the two
![]() -containing classes. The length-filtered datasets are generally
too small to be of practical use however (see
-containing classes. The length-filtered datasets are generally
too small to be of practical use however (see ![]() in
Table 3.1).
in
Table 3.1).
 |
At this point we should note that the filtering of the datasets applied here does not unfairly bias the prediction. The prediction accuracies quoted here are valid for any query sequence longer than a prescribed number of residues. The same is not true for datasets of protein domains with idealised class assignments (discussed above); in this case it is impossible to filter out borderline query sequences without first knowing their structures.