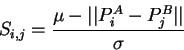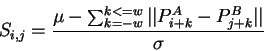| advertisement: compare things at compare-stuff.com! |
At each position in the query and library sequences, various properties and
quantities (discussed in Section 4.1.1) can be calculated from
the multiple sequence alignment. These can be combined into a property
vector ![]() (although initially we will use only one property at a time,
i.e.
(although initially we will use only one property at a time,
i.e. ![]() will be one dimensional). For query sequence A and library
sequence B, the individual vectors corresponding to residues
will be one dimensional). For query sequence A and library
sequence B, the individual vectors corresponding to residues ![]() and
and ![]() are designated
are designated ![]() and
and ![]() respectively. The standard dynamic
programming alignment algorithm requires a measure of suitability,
respectively. The standard dynamic
programming alignment algorithm requires a measure of suitability,
![]() for each residue
for each residue ![]() in the query sequence (A) at each position
in the query sequence (A) at each position
![]() in the library sequence (B), as described in more detail in
Section 1.2.1. No penalties are made for terminal gaps. In
order to align using property vectors, the value of
in the library sequence (B), as described in more detail in
Section 1.2.1. No penalties are made for terminal gaps. In
order to align using property vectors, the value of ![]() is calculated
as the normalised and negated Euclidean distance between property vectors
as follows:
is calculated
as the normalised and negated Euclidean distance between property vectors
as follows:
In order to measure local similarity, ![]() is calculated over a small
local window of
is calculated over a small
local window of ![]() residues each side of the central residue as follows:
residues each side of the central residue as follows:
To speed up the alignments of longer sequences, only values of ![]() within a certain distance of the central diagonal are calculated. This
distance (also, unfortunately, called a window) is set to
within a certain distance of the central diagonal are calculated. This
distance (also, unfortunately, called a window) is set to
![]() . Thus when
. Thus when ![]() , a maximum shift of more
than 50 residues between the two sequences is not possible. The
restriction could be removed at a later stage in the development or
application of the method.
, a maximum shift of more
than 50 residues between the two sequences is not possible. The
restriction could be removed at a later stage in the development or
application of the method.
Gap open and gap extension penalties of 3 and 0.5 respectively were used unless otherwise stated. These parameters are usually optimised empirically according to the requirements of a particular method. In this case, gap penalties were set early on in this investigation on the basis of a few trials, with the intention to refine them later.
Many threading methods use the dynamic programming algorithm in various forms, including local alignment[Jones et al., 1992], global alignment[Bowie et al., 1990,Matsuo & Nishikawa, 1995], and the so-called global-local alignment[Fischer & Eisenberg, 1996,Rice & Eisenberg, 1997]. These protocols basically differ in the scoring of terminal gaps and the extent of the alignment. The processing of scores in fold recognition is something of a black art. Theoretical proof exists to show that the scores from local alignments follow a Poisson-like distribution from which reasonable estimates of biological significance can be drawn[Henikoff, 1996,Bryant & Altschul, 1995, for reviews]. The widely used sequence database searching methods BLAST[Altschul et al., 1990], FASTA[Pearson, 1990] and Smith-Waterman[Smith & Waterman, 1981] all use local alignments. However, the distributions of global alignment scores are less well understood. Some methods use the global method to generate the alignment, and then calculate an energy score based on mounting the query sequence onto the library structure[Matsuo & Nishikawa, 1995], thus avoiding the direct use of the global score. Using global or local scores, Z-scores for each query-library pair can be calculated independently using scores from the alignments of randomised sequences[Rice & Eisenberg, 1997].
We found, however, that the global alignment algorithm[Needleman & Wunsch, 1970] gave the best results when combined with a simple score normalisation step. Before the calculation of Z-scores, the dynamic programming score is divided by the sum of the lengths of the two protein sequences. Without this correction, longer alignments (from longer library sequences) rank higher than they should.