| advertisement: compare things at compare-stuff.com! |
Exhaustive tests of the specific detection of remote homologues by standard sequence alignment methods have shown that optimum gap penalties are dependent upon the choice of amino acid substitution matrix[Pearson, 1995,Henikoff, 1996]. In this work, substitution matrices are not used; suitability is calculated from the Euclidean distances between property vectors. We investigate the possibility that the alignment of raw query sequences with structure-mapped library sequences requires different gap penalties.
Using a single set of mapped library sequences (sequence:structure ratio 4:1) and a control set of raw library sequences, full fold recognition trials (27 queries, 82 library domains) were performed with varying gap penalties. A total of 35 combinations of gap open (1, 2, 3, 4, 5, 6, 12) and gap extension (0.1, 0.3, 0.5, 1.0, 2.0) penalties were used. The best overall fold recognition performance was obtained using gap parameters of (3,0.5), (2,0.5) and (3,0.3); these trials used unmapped sequences. This agrees with the prior and undocumented optimisation of gap penalties (with different assessment criteria) from which we chose the default values of (3,0.5). The best fold recognition results using mapped sequences were obtained using gap penalties of (1,1), (2,1) and (1,2). These rankings were all worse than the best rankings using raw data, but it is interesting to note that the gap penalties are different.
Turning now to alignment quality, we observe that the successful gap
penalty parameters have low extension penalties. The top combinations for
the raw alignments are (1,0.1), (3,0.1) and (2,0.1), and for the mapped
alignments they are basically the same: (2,0.1), (3,0.1) and (1,0.1). The
alignment quality is not found to be improved by the use of mapping with
these optimal gap penalties, unlike the marginal improvement seen with
![]() doubly wound folds in Section 5.5.3.
doubly wound folds in Section 5.5.3.
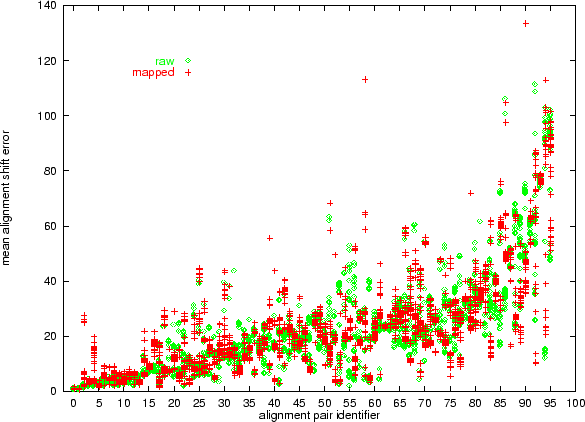
Figure 5.14 shows the mean alignment shift for each of the 35 gap penalty combinations for alignments of raw and mapped sequence information. Even with a wide range of gap penalties, the quality of the better alignments is surprisingly invariant, whilst the poorer alignments exhibit more diversity. Once again the results show that mapped alignments can be either consistently better or worse than unmapped alignments.
 |