| advertisement: compare things at compare-stuff.com! |
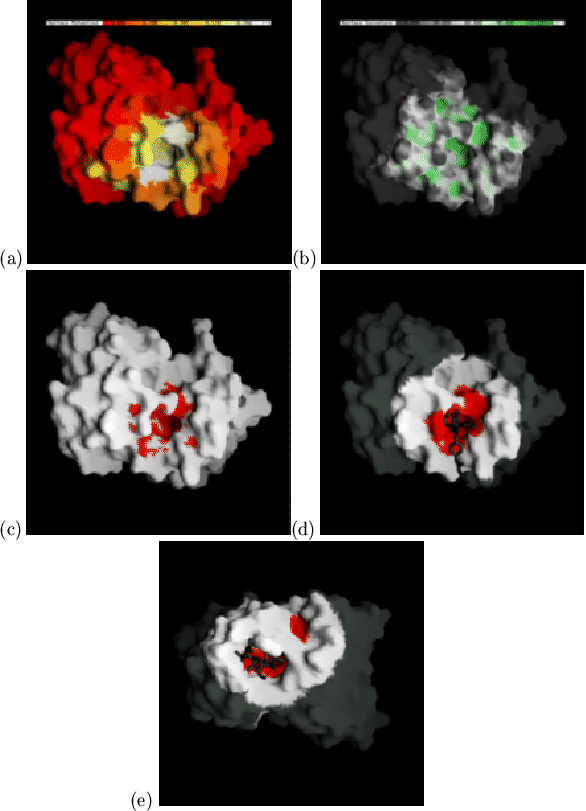
Having investigated relationships between antibody sequences, antigen contacts, antigen type and combining site topography, we applied these results by predicting the antigen-binding pockets of antibody structures. This is particularly relevant for antibodies whose sequences are known and from which a model can be constructed. From the model we aim to predict those residues most likely to be involved in antigen contact, which can then be subjected to site-directed mutagenesis to test both theory and model.
We have developed a rapid and simple method which uses only average contact
and surface shape information and test it by application to the complexed
crystal structures (all antigen-contact data from the antibody being
predicted are excluded). Firstly the mean burial (by antigen) data,
![]() , for each residue is projected onto the molecular surface
of the antibody (Figure 2.7(a)). Figure 2.7(b) shows
the same antibody with the surface coloured according to its curvature,
, for each residue is projected onto the molecular surface
of the antibody (Figure 2.7(a)). Figure 2.7(b) shows
the same antibody with the surface coloured according to its curvature,
![]() , as calculated by the program GRASP[Nicholls et al.,
1991]. This
alternative description of curvature was adopted for purely practical
reasons and allows easy application of the technique by anyone with access
to GRASP
, as calculated by the program GRASP[Nicholls et al.,
1991]. This
alternative description of curvature was adopted for purely practical
reasons and allows easy application of the technique by anyone with access
to GRASP![]() . To find the probable
contact surface we calculate
. To find the probable
contact surface we calculate
![]() , which combines the
measure of concavity with the probability of antigen contact. An arbitrary
cutoff of
, which combines the
measure of concavity with the probability of antigen contact. An arbitrary
cutoff of
![]() was chosen by visual inspection
(using just one complex), and surface points satisfying this condition are
coloured red (Figure 2.7(c)). This red surface is patchy and
discontinuous, so neighbouring patches are merged and disconnected patches
are eliminated using a dilation/erosion procedure[Delaney, 1992]
developed within GRASP. The red surfaces are dilated five times, eroded eight
times and dilated again three times (each by 1Å). The binding pocket
prediction is the resultant smooth red patch (Figure 2.7(d)).
was chosen by visual inspection
(using just one complex), and surface points satisfying this condition are
coloured red (Figure 2.7(c)). This red surface is patchy and
discontinuous, so neighbouring patches are merged and disconnected patches
are eliminated using a dilation/erosion procedure[Delaney, 1992]
developed within GRASP. The red surfaces are dilated five times, eroded eight
times and dilated again three times (each by 1Å). The binding pocket
prediction is the resultant smooth red patch (Figure 2.7(d)).
 |
The `worst case' accuracy, ![]() , and `fraction correct' accuracy,
, and `fraction correct' accuracy, ![]() ,
for predicting antigen contacting residues by this protocol with each of
the 26 complexed crystal structures are calculated as follows:
,
for predicting antigen contacting residues by this protocol with each of
the 26 complexed crystal structures are calculated as follows: