| advertisement: compare things at compare-stuff.com! |
The hydrophobic core residues of proteins are more conserved than non-core
residues due to the evolutionary constraints of sidechain packing in the
core. Conservation, therefore, is also a sequence feature
which clusters in three dimensions in protein structures. The measure of
conservation ![]() , introduced in Section 4.3.3, can be mapped
in the same manner as hydrophobicity in the previous section.
, introduced in Section 4.3.3, can be mapped
in the same manner as hydrophobicity in the previous section.
As mentioned previously, however, functional constraints restrict the
freedom of amino acid substitutions (in the active site of a protein for
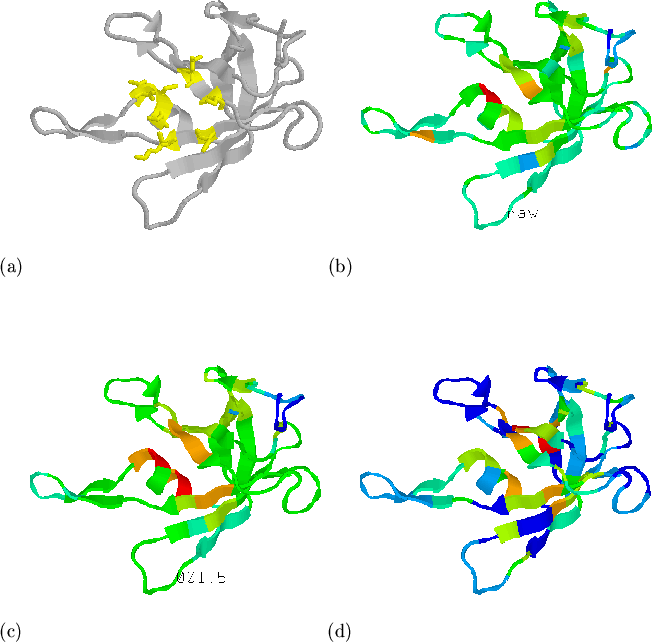
example), and hence affect conservation. In Figure 5.11 we
investigate the distribution of conserved residues in the structure of
domain 1knb01[Bruns & Karplus, 1995] from spinach ferredoxin reductase.
Figure 5.11(a) shows the active site residues (from the SITE
records in the PDB file) in yellow. Figures 5.11(b) and (c)
show the raw and mapped conservation measure ![]() , respectively. Compared
to the mapping of hydrophobicity shown in (d), the distribution of
conserved residues is more a consequence of function than structure. Since
function is not necessarily conserved between structures with similar
folds, the use of sequence conservation measures as described here may not
help to detect analogues. The identification of active
sites[Zvelebil et al.,
1987,Peters et al.,
1996,Laskowski et al.,
1996,Cabral et al.,
1996] using this technique
remains an interesting possibility.
, respectively. Compared
to the mapping of hydrophobicity shown in (d), the distribution of
conserved residues is more a consequence of function than structure. Since
function is not necessarily conserved between structures with similar
folds, the use of sequence conservation measures as described here may not
help to detect analogues. The identification of active
sites[Zvelebil et al.,
1987,Peters et al.,
1996,Laskowski et al.,
1996,Cabral et al.,
1996] using this technique
remains an interesting possibility.
 |