| advertisement: compare things at compare-stuff.com! |
Whilst the best results using raw information were obtained using secondary
structure prediction and hydrophobicity combined
(Table 4.9), the initial experiments using mapping,
for simplicity, use just hydrophobicity (Kyte and Doolittle).
Figure 5.12 shows the results of 17 different fold
recognition trials conducted using independent mappings employing different
ratios of sequence to structure information. According to the mean adjusted
rank measure (lower half of Figure 5.12(a)), fold recognition
performance does not improve beyond the baseline value of
![]() obtained using alignments of the same unmapped
information. In fact its lowest levels (ratios between 4.5:1 and 5.5:1)
appear to be about equal to this value. The poor performance for low
ratios is expected since the maps are heavily biased towards structure.
obtained using alignments of the same unmapped
information. In fact its lowest levels (ratios between 4.5:1 and 5.5:1)
appear to be about equal to this value. The poor performance for low
ratios is expected since the maps are heavily biased towards structure.
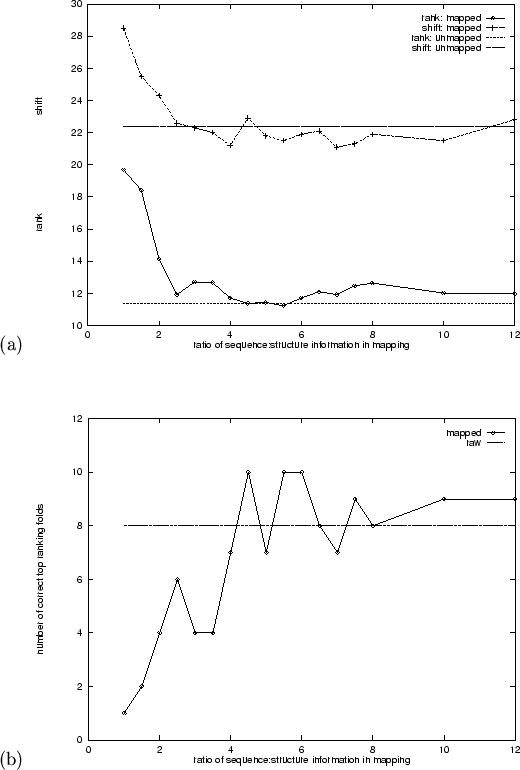 |
The results for alignment quality are more promising. The upper part of
Figure 5.12(a) shows that the mean alignment shift error,
![]() , is lower than the baseline value from alignments of
unmapped information (
, is lower than the baseline value from alignments of
unmapped information (
![]() ) for nearly all mappings with
sequence:structure ratios greater than 3:1.
) for nearly all mappings with
sequence:structure ratios greater than 3:1.
The number of correct top predictions, ![]() , from this experiment is shown
in Figure 5.12(b). Values of
, from this experiment is shown
in Figure 5.12(b). Values of ![]() occur three times in the
region of the plot corresponding to the minima in
Figure 5.12(a), suggesting that the mapping does improve the
specificity of fold recognition. However these data are very noisy (this
is expected using discrete integer measures) and a result of
occur three times in the
region of the plot corresponding to the minima in
Figure 5.12(a), suggesting that the mapping does improve the
specificity of fold recognition. However these data are very noisy (this
is expected using discrete integer measures) and a result of ![]() occurs
between two of these top scoring trials. We repeated this experiment on a
larger dataset (78 queries, 197 library domains) and obtained fewer correct
hits than the unmapped sequence control (23) using sequence to structure
ratios 4:1, 4.5:1, 5:1. Fold recognition is not improved using mapped
sequences, with the methods presented here.
occurs
between two of these top scoring trials. We repeated this experiment on a
larger dataset (78 queries, 197 library domains) and obtained fewer correct
hits than the unmapped sequence control (23) using sequence to structure
ratios 4:1, 4.5:1, 5:1. Fold recognition is not improved using mapped
sequences, with the methods presented here.
The problem with the results presented here is that we are trying to compare results from multiple mappings and alignments with a single result from alignments of unmapped data. From Figure 5.12 it is clear that the results are prone to noise. It is not unreasonable to suppose that the alignments of unmapped sequence information are not perfectly stable. Therefore replicate fold recognition trials should be performed with both mapped and unmapped data to assess the spread of likely results. For the mapping protocol this is easy; the self-organising mapping algorithm is non-deterministic and suitable for replication. Alignments of unmapped information are deterministic and cannot be repeated. Furthermore, since a full fold recognition trial, including map generation, takes a few hours, the number of repeat experiments cannot be very large. Alternative approaches were devised to investigate the results more thoroughly.